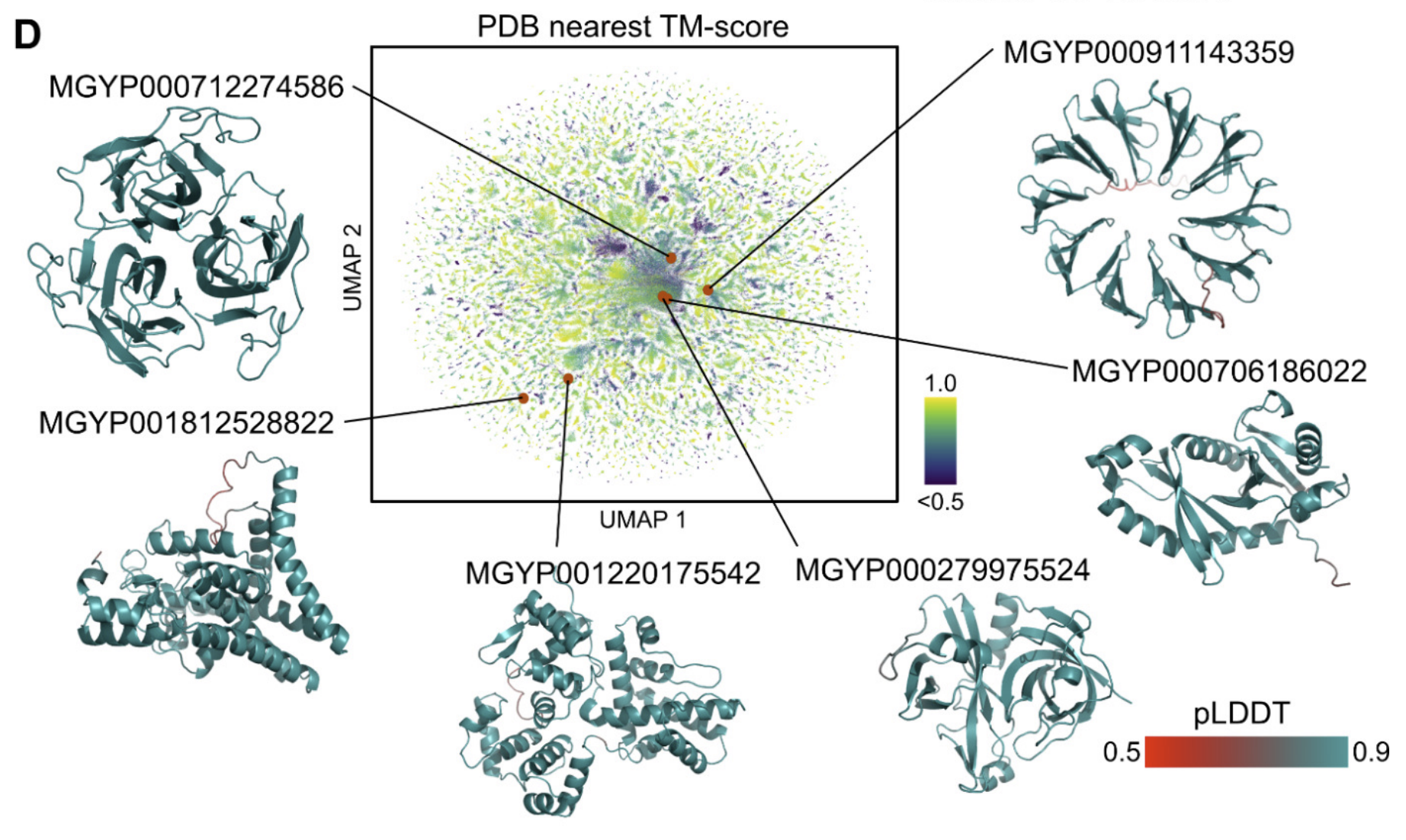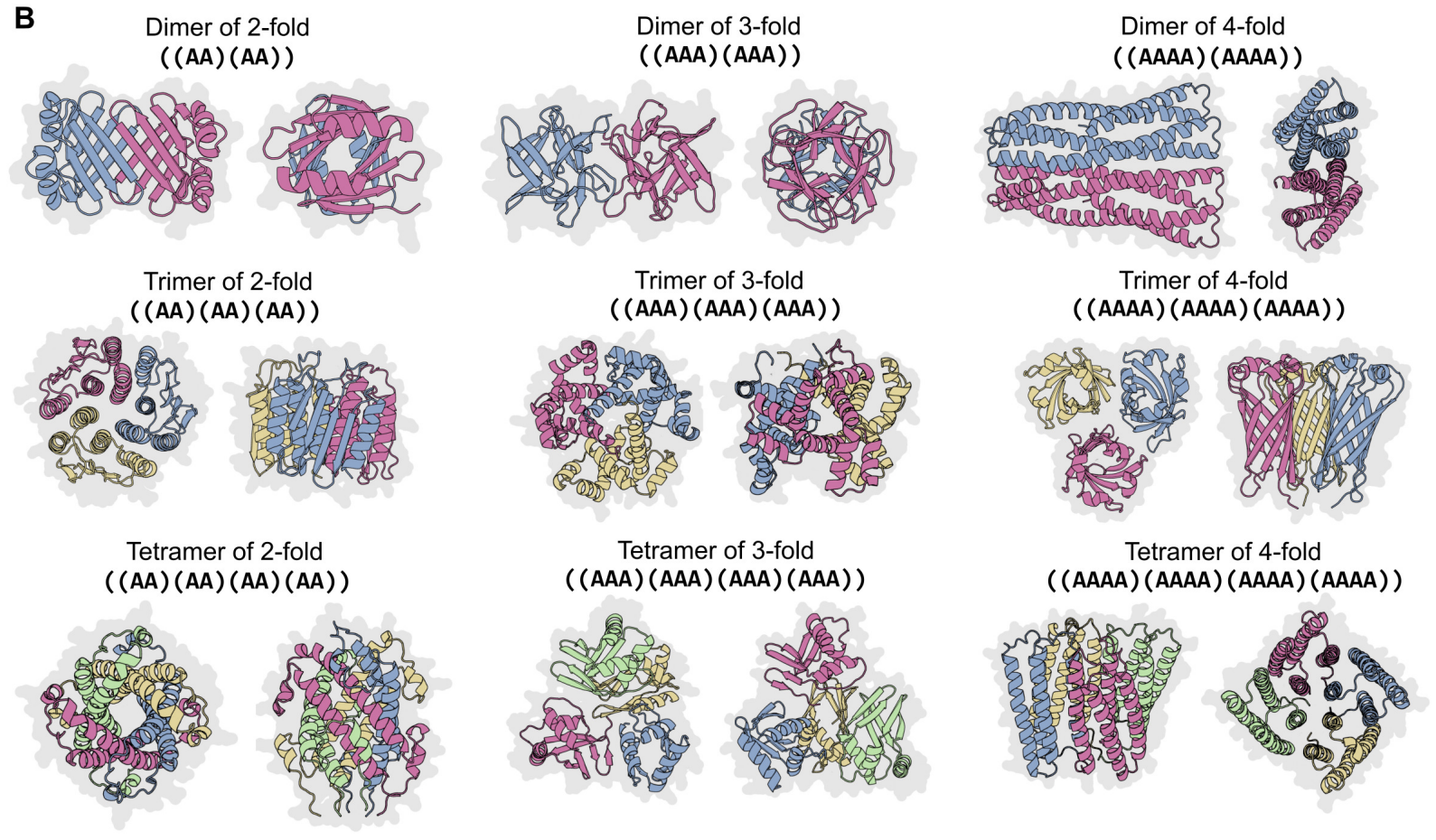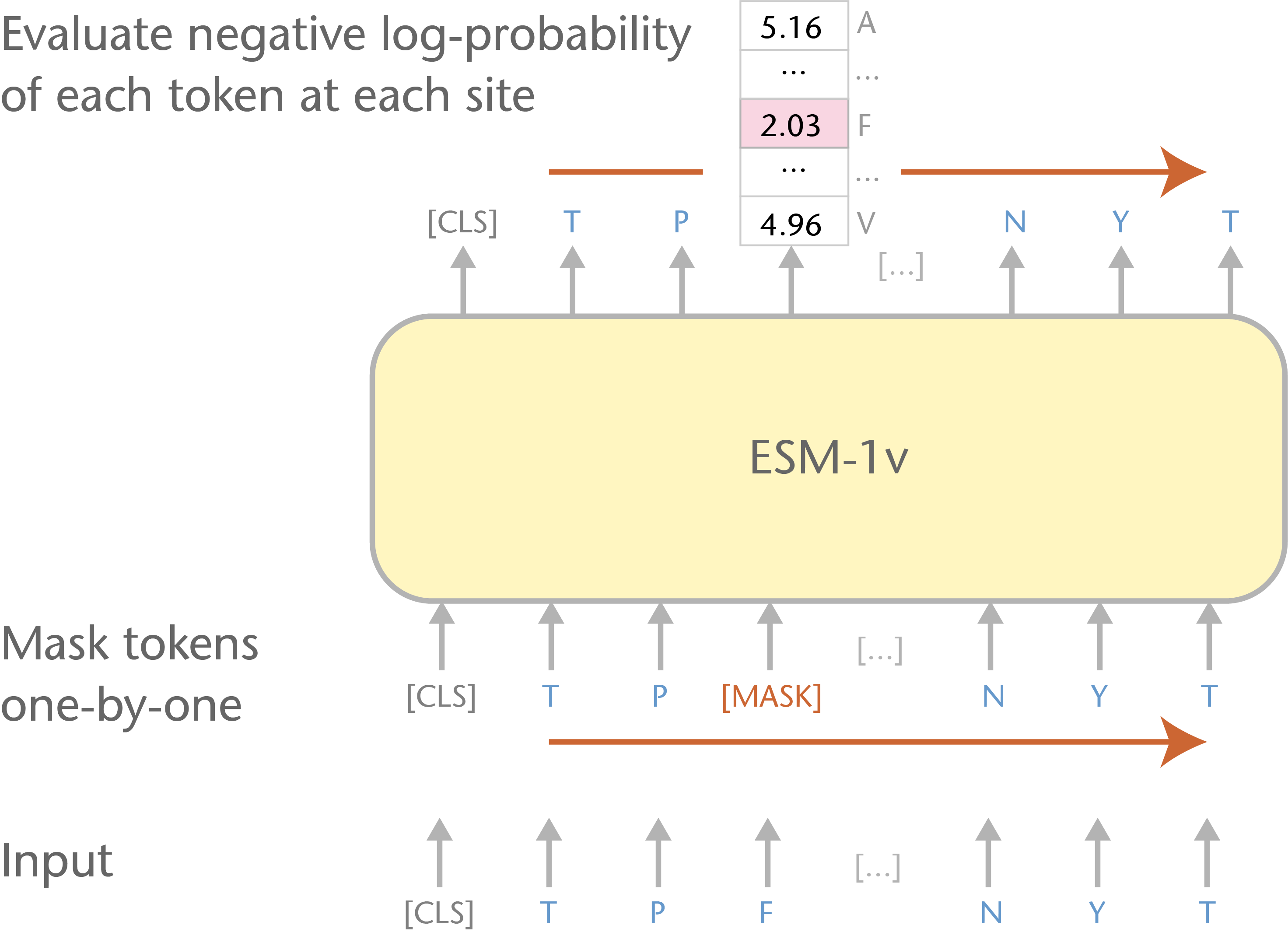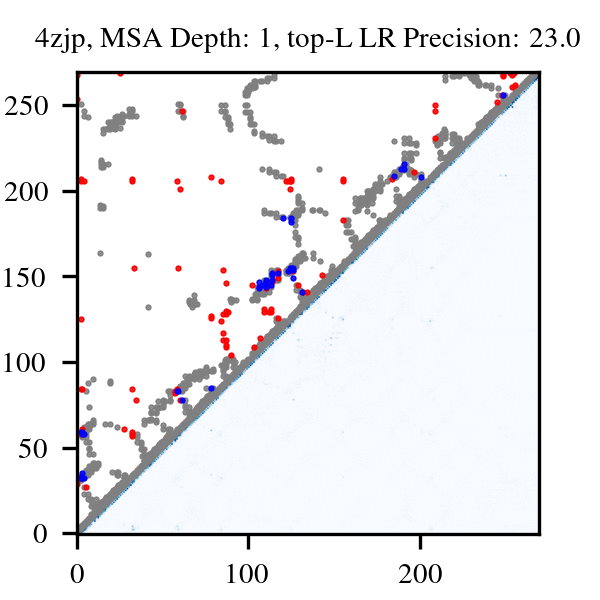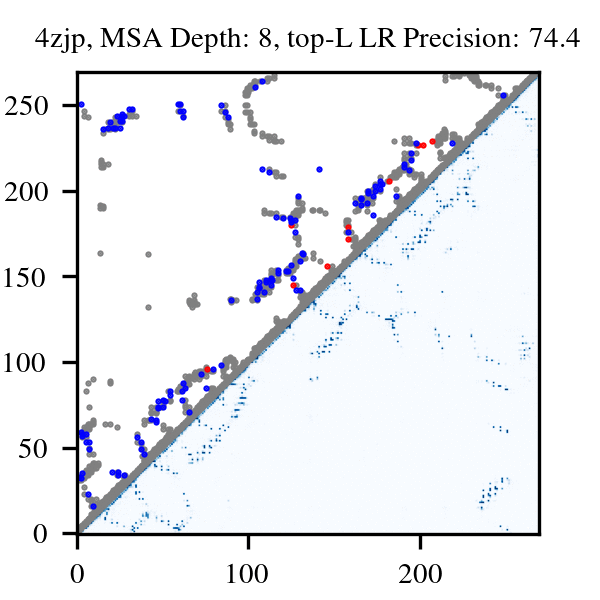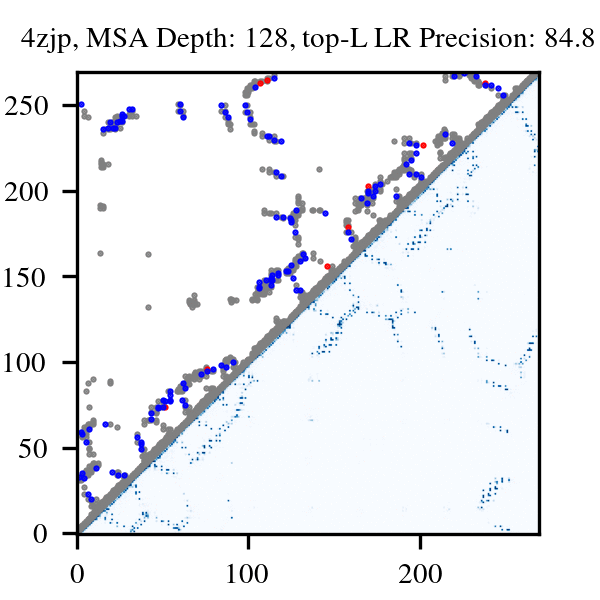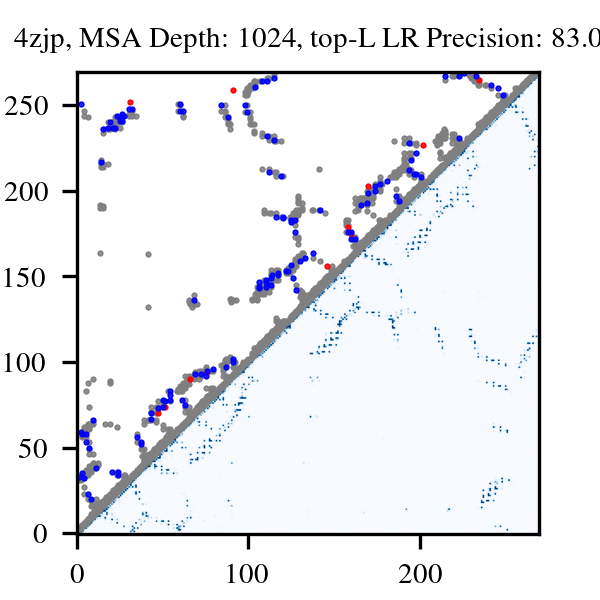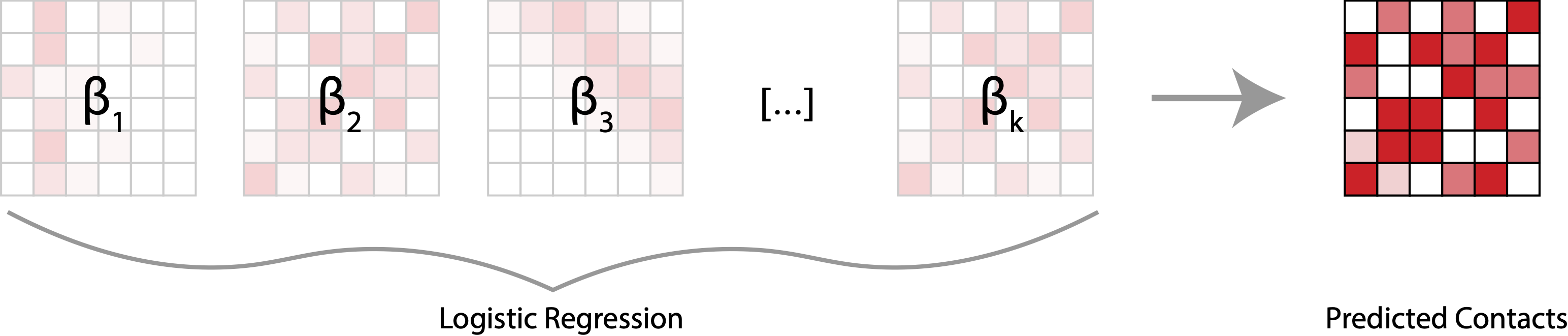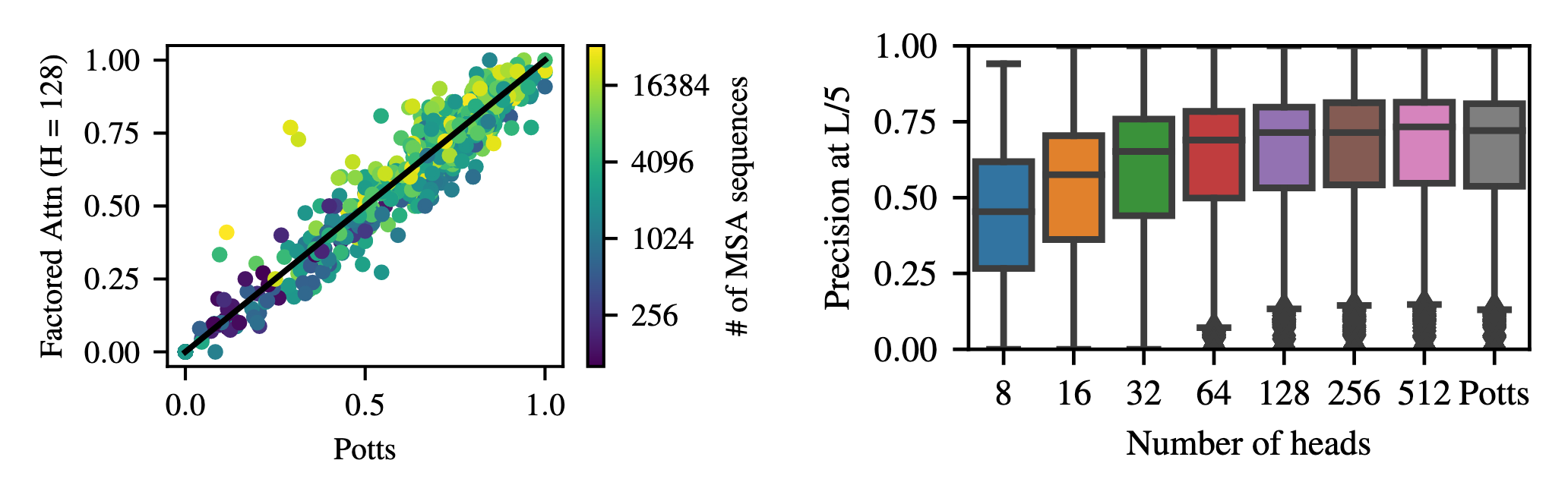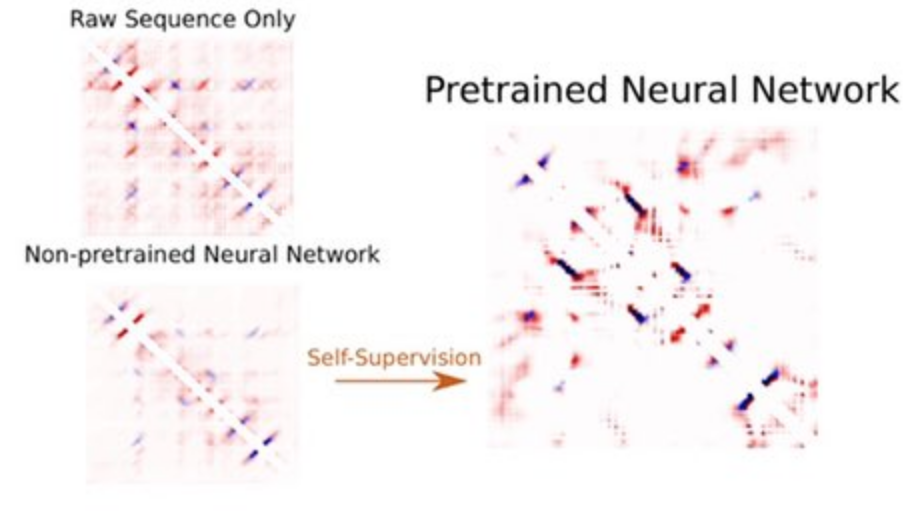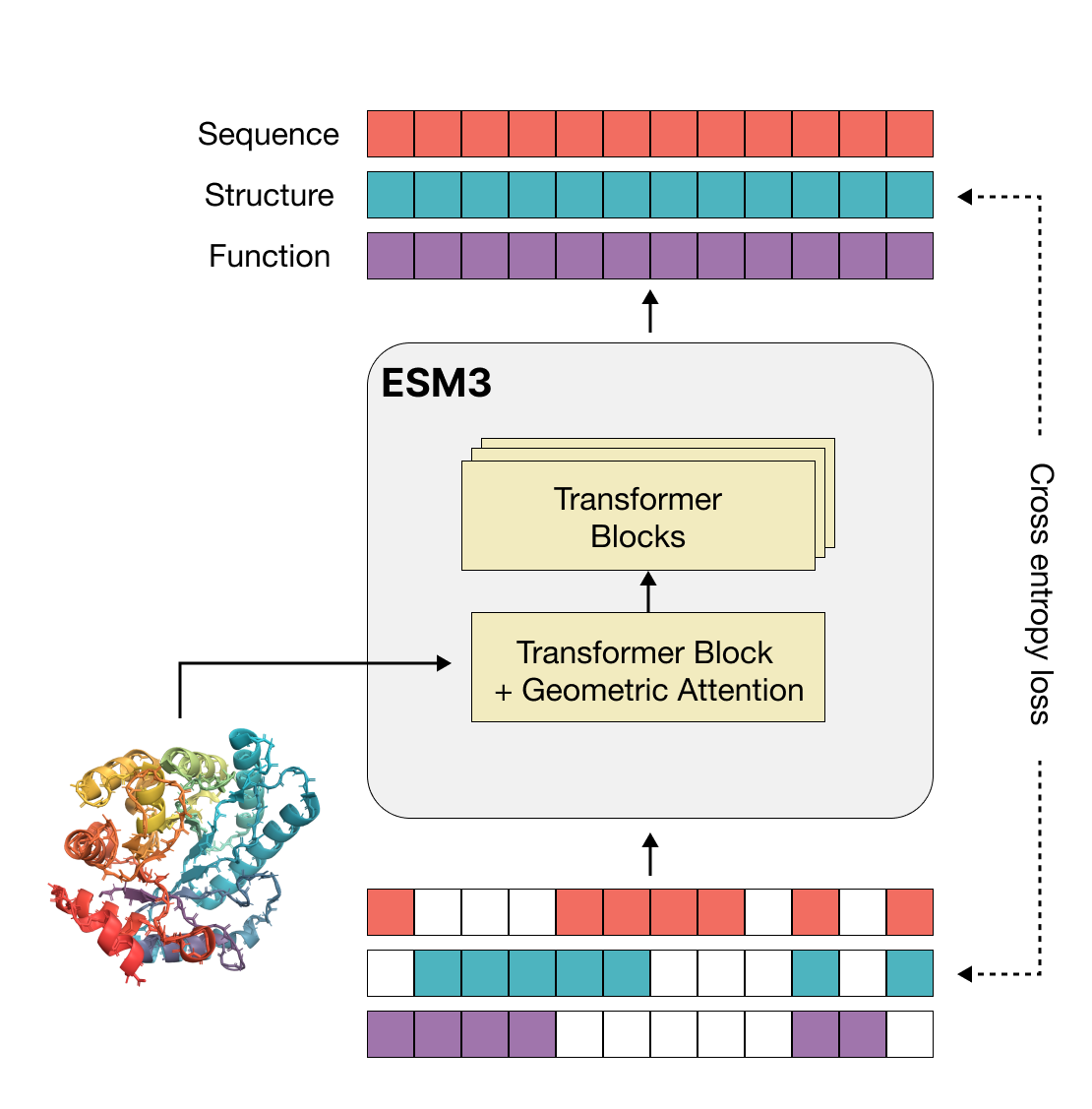
More than three billion years of evolution have produced an image of biology encoded into the space of natural proteins. Here we show that language models trained on tokens generated by evolution can act as evolutionary simulators to generate functional proteins that are far away from known proteins. We present ESM3, a frontier multimodal generative language model that reasons over the sequence, structure, and function of proteins. ESM3 can follow complex prompts combining its modalities and is highly responsive to biological alignment. We have prompted ESM3 to generate fluorescent proteins with a chain of thought. Among the generations that we synthesized, we found a bright fluorescent protein at far distance (58% identity) from known fluorescent proteins. Similarly distant natural fluorescent proteins are separated by over five hundred million years of evolution.